
What is the Carrier Screening for Recessive Disorders test?
The carrier screening is a genetic test which identifies mutations in recessive disease-related genes, developed for couples in family planning.
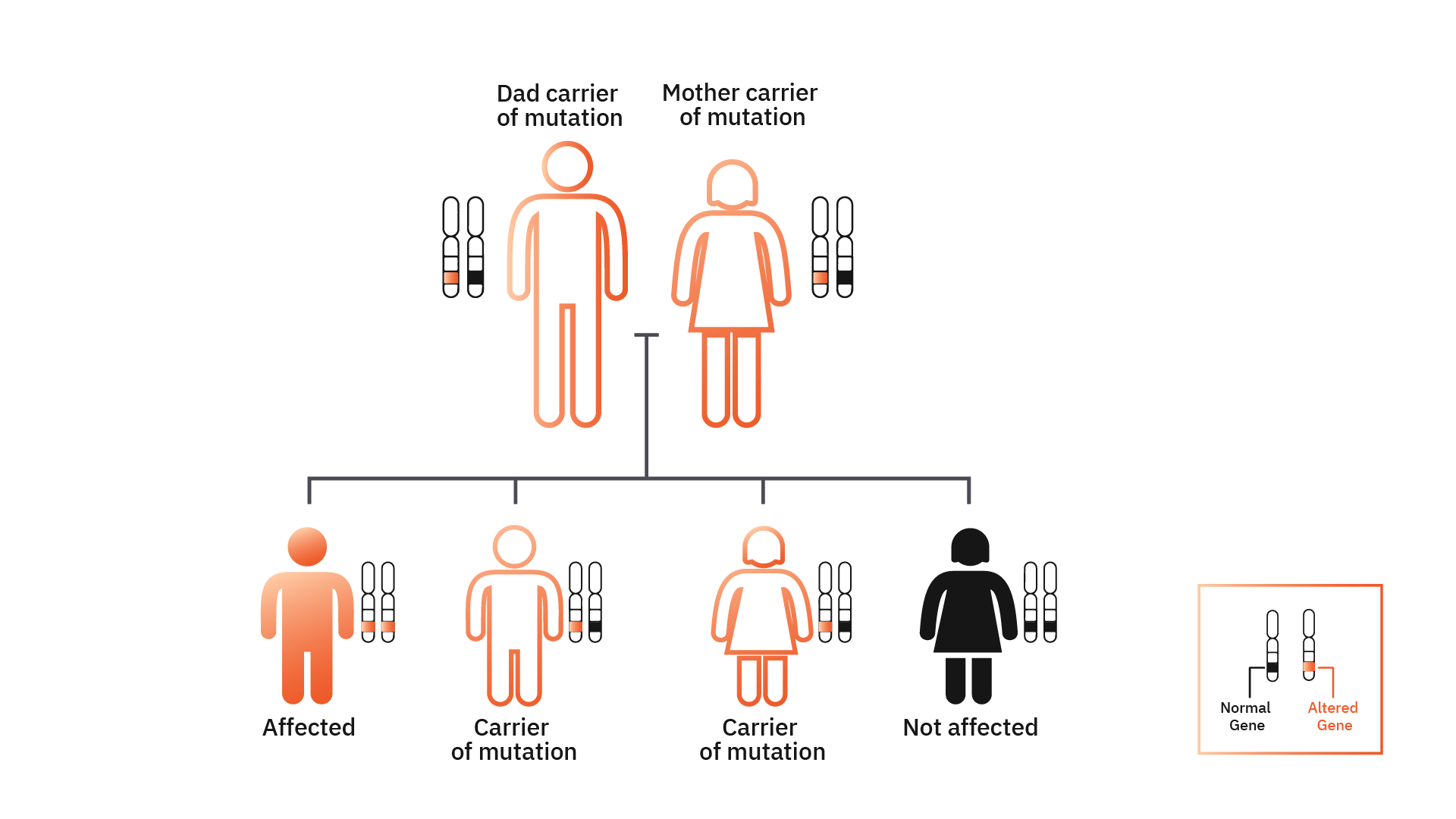
Understand how diseases with recessive inheritance are shared in a family.
Does the test need to be requested by a physician?
The screening test does not need to be requested by a physician, but, it is recommended that the results are always presented to a physician who will provide directions to the couple concerning the family planning options and complementary tests.
What to expect from this test result?
If the two members of the couple are tested, the result may provide information concerning the risk of having a child with a recessive genetic condition.
If the result indicates that the couple have a mutation in the same gene, the risk of having a child with a genetic disease is of at least 25%. In this case the couple may decide for an in vitro fertilization (IVF) and preimplantation genetic diagnosis (PGD).
When and who can submit to the test?
- Couples in family planning
- People with increased risk for recessive disorders
- Reproduction clinics working with sperm and egg donors
This test must be used as a genetic counseling tool for people with increased risk of being carriers of mutations that lead to recessive disease.
Particularly indicated for consanguineous couples or for populations with increased risk for recessive diseases, such as the Jewish population, particularly of Ashkenazi inheritance, but not limited to it, extending to other populations such as Sephardic and Mizrahi ones, for example.
Why are some couples at major risk of having children with genetic diseases?
We all have two copies of each gene, a maternal copy and a paternal one. Most of the genetic diseases are recessive. This means that for the disease to manifest, it’s necessary that the two gene copies (maternal and paternal) are mutated (altered). By inheriting just one copy of a mutated gene (maternal or paternal copy), the person will only be a mutation carrier, but will not present the disease.
Any couple may have a child with a genetic disease, but such risk is increased for consanguineous couples (having a degree of relatedness), certain ethnic groups with increased risk for specific genetic conditions (e.g.: Ashkenazi Jew), or people with family history of recessive genetic disease such as cystic fibrosis and sickle-cell anemia.
These couples present an increased risk of being carriers of mutations in the same genes. For this reason, it’s important that couples under such conditions seek a specialist to become aware of their risk before starting a family and, currently, there are already genetic tests able to assist them.
Main Diseases Analyzed
- Fanconi Anemia C (FANCC)
- Dihydrolipoamide Dehydrogenase Deficiency (DLD)
- Family Dysautonomia – Hereditary Sensory and Autonomic Neuropathy (ELP1)
- Canavan Disease (ASPA)
- Niemann-Pick A/B Disease (SMPD1)
- Tay-Sachs Disease (HEXA)
- Maple Syrup Disease (BCKDHB)
- Cystic Fibrosis (CFTR)
- Glycogenosis 1A – Von Gierke (G6PC1)
- Hyperinsulinemic Hypoglycemia 1 (ABCC8)
- Nemaline Myopathy 2 (NEB)
- Mucolipidosis IV (MCOLN1)
- Bloom Syndrome (BLM)
- Gaucher Syndrome (GBA)
- Usher Syndrome 1D/1F (PCDH15)
- Usher Syndrome 3A (CLRN1)
Other diseases analyzed in this exam
- Other diseases analyzed in this test
- Abetalipoproteinemia
- Glutaric Acidemia I
- Isovaleric Acidemia
- Propionic Acidemia
- 3-Methylglutaconic Aciduria III
- Argininosuccinic Aciduria
- Methylmalonic Aciduria and Homocystinuria
- Methylmalonic Aciduria
- Corpus Callosum Agenesis and Peripheral Neuropathy
- Fanconi Anemia A
- Sickle-Cell Anemia and Thalassemia
- Argininemia
- Arthrogryposis, Intellectual Disability and Epilepsy
- Aspartylglucosaminuria
- Spastic Ataxia
- Ataxia-Telangiectasia
- Nephropathic Cystinosis
- Citrullinemia
- Combined Pituitary Hormone 2 Deficiency
- 3-Hydroxy-3-Methylglutaryl-CoA Lyase Deficiency
- Short Chain Acyl-Coa Dehydrogenase Deficiency
- Medium Chain Acyl-Coa Dehydrogenase Deficiency
- Very Long Chain Acyl-Coa Dehydrogenase Deficiency
- Biotinidase Deficiency
- Carbamoyl Phosphate Synthetase I Deficiency
- Carnitine Palmitoyltransferase I Deficiency
- Mitochondrial Complex III Deficiency Type 1
- Factor XI Deficiency
- Galactokinase Deficiency (Galactosemia)
- Holocarboxylase Synthetase Deficiency
- Lysosomal Acid Lipase Deficiency
- Multiple Sulfatase Deficiency
- Pyruvate Carboxylase Deficiency
- Mitochondrial Trifunctional Protein Deficiency
- Vitamin E Deficiency
- Primary Systemic Carnitine Deficiency
- Spondylocostal Dysostosis 2
- Skeletal Dysplasia
- Congenital Dyskeratosis 5
- Congenital Muscular Dystrophy and Brain and Eye Anomalies A3
- Congenital Muscular Dystrophy and Brain and Eye Anomalies A4
- Congenital Muscular Dystrophy and Brain and Eye Anomalies A5
- Merosin-Deficient Congenital Muscular Dystrophy 1A
- Limb-girdle Muscular Dystrophy 2A
- Limb-girdle Muscular Dystrophy 2B
- Limb-girdle Muscular Dystrophy 2C
- Limb-girdle Muscular Dystrophy 2D
- Limb-girdle Muscular Dystrophy 2E
- Limb-girdle Muscular Dystrophy 2F
- Congenital Glycosylation Disorder (CDG) IA
- Congenital Glycosylation Disorder (CDG) IB
- Congenital Glycosylation Disorder (CDG) IC
- Peroxisome Biogenesis Disorder 1A/1B
- Peroxisome Biogenesis Disorder 3A/3B
- Peroxisome Biogenesis Disorder 4A/4B
- Peroxisome Biogenesis Disorder 5A/5B
- Peroxisome Biogenesis Disorder 6A/6B
- Peroxisome Biogenesis Disorder 9B
- Infantile Sialic Acid Storage Disorder
- Gaucher Disease
- Krabbe Disease
- Niemann-Pick Disease C1
- Niemann-Pick Disease C2
- Sandhoff Disease
- Segawa Disease
- Wilson Disease
- Maple Syrup Disease
- Renal Polycystic Disease
- Glycine Encephalopathy
- Junctional Epidermolysis Bullosa
- Phenylketonuria
- Galactosemia
- Gangliosidosis GM1 I
- Glycogenosis 1B/1C
- Glycogenosis 2 (Pompe)
- Glycogenosis 3 (Forbes)
- BH4-Deficient Hyperphenylalaninemia A
- Primary Hyperoxaluria I
- Primary Hyperoxaluria II
- Primary Hyperoxaluria III
- Congenital Lipoid Adrenal Hyperplasia
- Congenital Adrenal Hyperplasia
- Hypophosphatasia
- Hyperinsulinemic Hypoglycemia 2
- Homocystinuria
- Congenital Ichthyosis 1
- Combined Severe Immunodeficiency (SCID)
- Fructose Intolerance
- Metachromatic Leukodystrophy
- Megalencephalic Leukoencephalopathy and Subcortical Cysts 1
- Ceroid Lipofuscinosis (CLN) 1
- Ceroid Lipofuscinosis (CLN) 2
- Ceroid Lipofuscinosis (CLN) 3
- Ceroid Lipofuscinosis (CLN) 4A/6
- Ceroid Lipofuscinosis (CLN) 5
- Ceroid Lipofuscinosis (CLN) 8
- Lysosomal Mannosidase Alpha B
- Nonaka Myopathy
- Mucolipidosis IIA/IIB
- Mucolipidosis IIIC
- Mucopolysaccharidosis 1H (Hurler-Scheie)
- Mucopolysaccharidosis 3A (Sanfilippo)
- Mucopolysaccharidosis 3B (Sanfilippo)
- Mucopolysaccharidosis 3C (Sanfilippo)
- Osteopetrosis 1
- Spastic Paraplegia (SPG) 15
- Pycnodysostosis
- Retinitis Pigmentosa 59
- Alport Syndrome
- Alstrom Syndrome
- Bardet-Biedl Syndrome (BBS) 1
- Bardet-Biedl Syndrome (BBS) 10
- Bardet-Biedl Syndrome (BBS) 12
- Bardet-Biedl Syndrome (BBS) 2
- Cockayne Syndrome A
- Cockayne Syndrome B
- Cohen Syndrome
- Ehlers-Danlos Syndrome VII
- Leigh Syndrome
- Meckel Syndrome 1
- Meckel Syndrome 2
- Pendred Syndrome 1
- Perrault Syndrome 1
- Nijmegen Breakage Syndrome
- Sjogren-Larsson Syndrome
- Usher Syndrome 1
- Usher Syndrome 1C
- Usher Syndrome 2A
- Ellis-van Creveld Syndrome
- Hydrolethalus Syndrome 1
- Nephrotic Syndrome 1
- Nephrotic Syndrome 2
- Neu-Laxova Syndrome 1
- Autoimmune Polyendocrine Syndrome and Reversible Metaphyseal Dysplasia I
- Smith-Lemli-Opitz Syndrome
- Deafness
- Tyrosinemia I
- Tyrosinemia II
- Congenital Amegakaryocytic Thrombocytopenia
- Cerebrotendinous Xanthomatosis
- Xeroderma Pigmentosum A
- Xeroderma Pigmentosum C
Mendelics Differentials

Pioneer in Brazil and in Latin America
The first and the largest laboratory focused on genomic analysis in Latin America.

More precise
results
The largest database of Brazilian and Latin American variants, with more than 100 thousand genetic tests conducted.

International Quality Certifications
The only Brazilian laboratory specialized in genetics with CAP, PALC and ISO, the main international certifications.

Agility to deliver the reports
Proprietary software with artificial intelligence for genetic data analysis, MIT award winner for innovation.
