
A Triagem de Portador de Mutações de Doenças Recessivas e Ligadas ao X é um teste genético que identifica variantes patogênicas (mutações) em genes relacionados a doenças recessivas.
O teste de triagem do portador pode ser feito por casais em planejamento familiar, casais consanguíneos, pessoas com risco aumentado e/ou com histórico familiar de doenças recessivas.
*Todo teste genético tem seus benefícios e limitações. Converse com um médico de confiança para entender as possibilidades de realizar um teste de triagem do portador.
Por que alguns casais têm maior risco de ter filhos com doenças genéticas?
A maioria dos genes humanos em nossas células estão em pares. Nós herdamos uma cópia do gene (alelo) materno e outra cópia do gene (alelo) paterno.
Para uma doença recessiva se manifestar, é necessário que as duas cópias do gene (alelos) estejam mutadas (alteradas). Quando uma pessoa tem apenas uma cópia do gene mutado (a cópia materna ou paterna), ela será chamada de portadora.
- Pessoas homozigotas para a mutação, isto é, com as duas cópias mutadas, manifestam a doença recessiva.*
- Pessoas heterozigotas para a mutação, isto é, com apenas uma cópia mutada, geralmente não têm sintomas.*
Assim, casais nos quais ambos os parceiros têm a mesma cópia (alelo) com mutação associada a uma doença recessiva têm maior chance de ter um filho afetado pela condição.
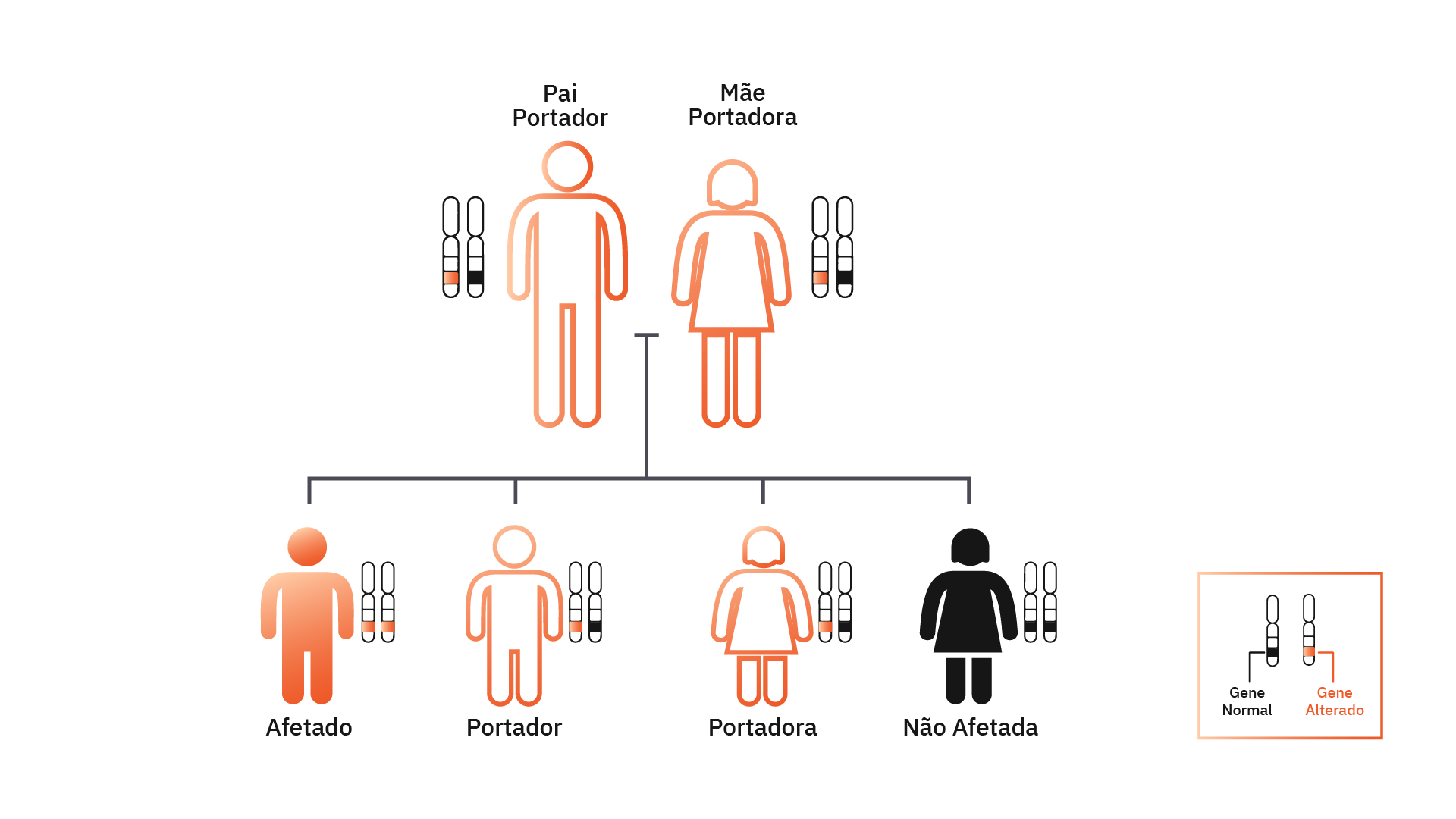
A imagem ilustra um casal de portadores, com 50% de chance de ter um filho(a) portador, 25% de chance de ter um filho(a) afetado pela doença e 25% de ter um filho(a) não afetado.
Isso não significa que um casal que não tem os alelos recessivos não possa ter um filho com a doença, visto que mutações novas (de novo) podem acontecer durante a formação do embrião.
Qualquer casal pode ter um filho com uma doença genética, mas esse risco é maior para:
- Casais consanguíneos: que possuem grau próximo de parentesco biológico e, portanto, tem maior chance de compartilharem os mesmos alelos
- Determinados grupos étnicos com risco aumentado de condições genéticas específicas (exemplo: Judeu Ashkenazi)
- Pessoas com histórico familiar de doença genética recessiva, como fibrose cística, talassemia-beta e anemia falciforme.
Pessoas que se encaixam nesses critérios podem procurar um médico e entender os benefícios e limitações de fazer um teste de triagem de portador.
*A manifestação de uma doença também depende de fatores ambientais e da interação desses com o componente genético de um indivíduo.
O teste de triagem de portador precisa ser solicitado por um médico?
Não. O teste de triagem de portador da Mendelics não precisa ser solicitado por um médico, mas reforçamos a importância que os resultados sejam sempre acompanhados por um especialista, que orientará o casal quanto às opções de planejamento familiar e exames complementares.
Todo teste genético tem benefícios e limitações. Converse com seu médico de confiança.
O que esperar do resultado deste exame?
Se os dois membros do casal forem testados, o resultado poderá fornecer informações sobre o risco de ter um filho com uma condição genética recessiva.
Se o resultado indicar que o casal é portador de mutação no mesmo gene, o risco de nascer uma criança com doença genética aumenta. Nesse caso, o casal pode optar por um fertilização in vitro (FIV) e diagnóstico genético pré‐implantacional (PGD).
Quando e quem pode realizar o teste de triagem de portador?
- Casais em planejamento familiar
- Casais consanguíneos
- Pessoas com risco aumentado para doenças recessivas, como população judaica, principalmente de herança Ashkenazi, mas não se limitando a ela, podendo se estender a outras populações como Sefaradim e Mizrahim, por exemplo.
- Clínicas de reprodução que trabalham com doadores de espermatozóides e óvulos
Este exame deve ser utilizado como ferramenta de aconselhamento genético para pessoas com risco aumentado de serem portadores de mutação para doenças recessivas.
Saiba mais sobre a Triagem de Portador de Mutações de Doenças Recessivas e Ligadas ao X
Principais doenças analisadas pelo teste de Triagem de Portador de Mutações de Doenças Recessivas e Ligadas ao X
- Anemia de Fanconi C (FANCC)
- Deficiência de Diidrolipoamida Desidrogenase (DLD)
- Disautonomia Familiar – Neuropatia Sensitiva e Autonômica Hereditária 3 (ELP1)
- Doença de Canavan (ASPA)
- Doença de Niemann-Pick A/B (SMPD1)
- Doença de Tay-Sachs (HEXA)
- Doença de Xarope de Bordo (BCKDHB)
- Fibrose Cística (CFTR)
- Glicogenose 1A – Von Gierke (G6PC1)
- Hipoglicemia Hiperinsulinêmica 1 (ABCC8)
- Miopatia Nemalínica 2 (NEB)
- Mucolipidose IV (MCOLN1)
- Síndrome de Bloom (BLM)
- Síndrome de Gaucher (GBA1)
- Síndrome de Usher (PCDH15)
- Síndrome de Usher (CLRN1)
Ficou com alguma dúvida? Entre em contato conosco.
Diferenciais da Mendelics

Pioneiro no Brasil e América Latina
Primeiro e maior laboratório focado em análise genômica da América Latina.

Resultados mais
precisos
Maior banco de dados de variantes brasileiras e latino americanas, com mais de 100 mil exames genéticos realizados.

Qualidade internacional certificada
Único laboratório brasileiro especializado em genética com CAP, PALC e ISO, principais certificações internacionais.

Agilidade na entrega dos laudos
Software proprietário com inteligência artificial para análise de dados genéticos, vencedor do prêmio MIT de inovação.
